SomaticSignatures 教程:使用 TCGA 突变数据做肿瘤 signature 分析
SomaticSignatures 是一个用于分析肿瘤单核苷酸变异(SNV / SNP)模式的 R 包,常见用途是从样本的突变上下文里提取潜在的 mutational signatures,用来辅助理解肿瘤发生、发展和演化过程。
这篇文章不讲太多理论,重点放在一个可直接复用的流程上:先把 TCGA 的 MAF 数据整理成 SomaticSignatures 需要的表格,再在 R 里评估 signature 数量、做 NMF 分解并输出图形结果。
开始前先确认
在跑这套流程前,建议先确认 3 件事:
- 你已经准备好了包含 SNP 信息的 MAF 或类似突变表
- 参考基因组版本和后续分析使用的
BSgenome包一致,这里示例使用hg38 - 样本数量不要太少,否则 signature 分解结果会比较不稳定
数据准备
SomaticSignatures 需要的核心字段并不多,至少要整理出下面这些列:
Samplechrstartrefalt
如果你下载的是 TCGA 的原始 MAF,可以先用 Python 做一遍清洗,只保留 SNP,并把字段改成后续 R 代码更容易直接使用的形式。
下面是一个最直接的预处理示例:
import pandas as pd
df = pd.read_csv("TCGA.CRC.mutect.maf.csv")
df.head()
"""
Hugo_Symbol Entrez_Gene_Id ... MC3_Overlap GDC_Validation_Status
0 UBE2J2 118424 ... True Unknown
1 RPL22 6146 ... True Unknown
2 TNFRSF9 3604 ... True Unknown
3 EXOSC10 5394 ... False Unknown
4 PTCHD2 57540 ... True Unknown
"""
df = df[df.Variant_Type=='SNP']
#首先筛选snp数据
newdf = df.iloc[:,[33,4,5,11,12]]
#挑选特定列
newdf.columns = ["Sample","chr", "start","ref", "alt"]
newdf.ref = newdf.ref.str[:1]
# 去除ref与alt一样的行,否则后续会出错
newdf = newdf[newdf.ref != newdf.alt]
#保存数据用于R分析
newdf.to_csv("TCGA.CRC.mutect.maf.filtered.csv",index=None)
这一步的目标很明确:把复杂的 MAF 表压缩成一个干净的 SNP 表,供 R 侧直接读入。
筛选Signatures
这里使用 R 做后续分析,因为 signature 提取和绘图都依赖 SomaticSignatures 生态。
先安装必要的包:
install.packages(c("SomaticSignatures","SomaticCancerAlterations","BSgenome.Hsapiens.UCSC.hg38","data.table"))
加载依赖:
suppressPackageStartupMessages(library(SomaticSignatures))
suppressPackageStartupMessages(library(SomaticCancerAlterations))
suppressPackageStartupMessages(library(BSgenome.Hsapiens.UCSC.hg38))
suppressPackageStartupMessages(library(data.table))
suppressPackageStartupMessages(library(ggplot2))
然后把清洗后的表读进来,并转换成 mutationContext 可接受的 VRanges:
df=fread('TCGA.CRC.mutect.maf.filtered.csv',data.table = F)
# ["Sample","chr", "start","end","ref", "alt"]
alls=as.character(unique(df$Sample))
df$study=df$Sample
sca_vr = VRanges(
seqnames = df$chr ,
ranges = IRanges(start = df$start,end = df$start+1),
ref = df$ref,
alt = df$alt,
sampleNames = as.character(df$Sample),
study=as.character(df$study))
接着生成突变上下文矩阵,并评估不同 signature 数量下的拟合效果:
sca_motifs = mutationContext(sca_vr, BSgenome.Hsapiens.UCSC.hg38)
head(sca_motifs)
# 对每个样本,计算 96 突变可能性的 比例分布情况
escc_sca_mm = motifMatrix(sca_motifs, group = "study", normalize = TRUE)
dim( escc_sca_mm )
table(colSums(escc_sca_mm))
head(escc_sca_mm[,1:4])
n_sigs = 5:15
gof_nmf = assessNumberSignatures(escc_sca_mm , n_sigs, nReplicates = 5)
save(gof_nmf,file = 'gof_nmf.Rdata')
load(file = 'gof_nmf.Rdata')
# 这个 assessNumberSignatures 步骤耗时很严重。
pdf("plotNumberSignatures.pdf",width=18, height=7)
plotNumberSignatures(gof_nmf)
dev.off()
这里最耗时的步骤通常是:
assessNumberSignatures(escc_sca_mm , n_sigs, nReplicates = 5)
如果样本数量多,或者你把 nReplicates 设得更高,运行时间会明显增加。
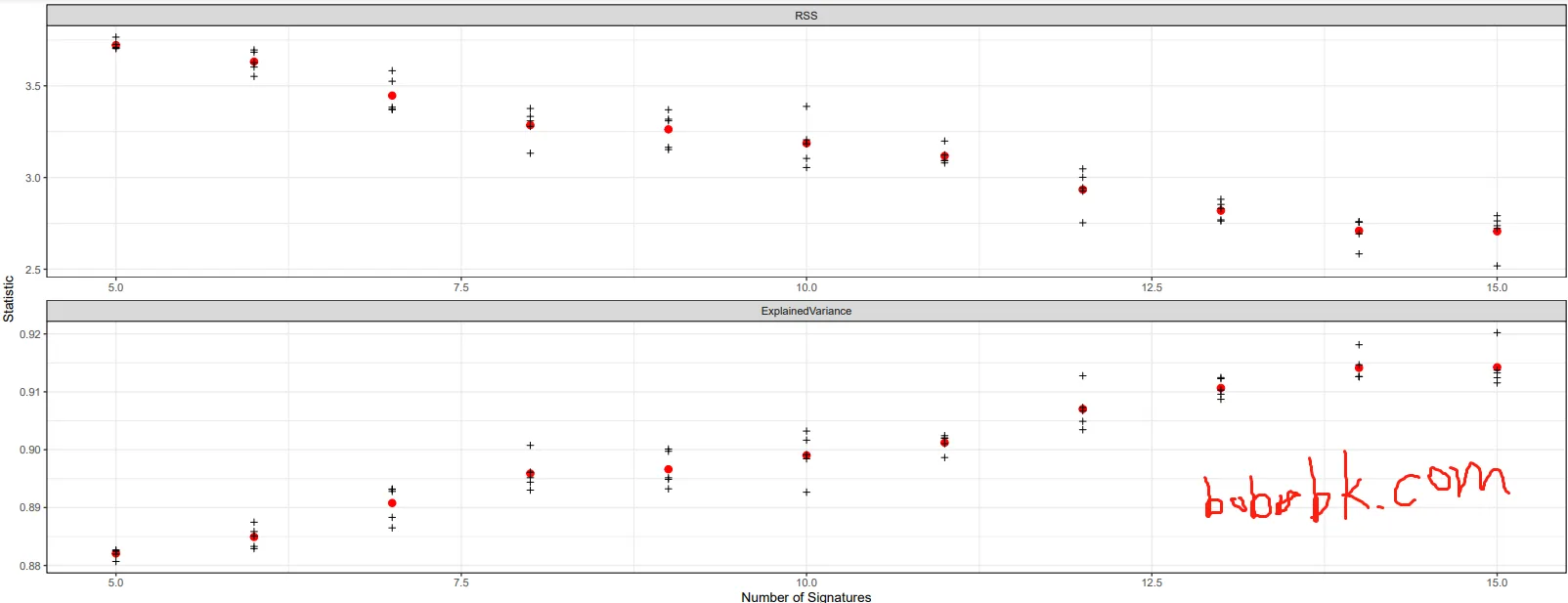
结果示例如下:
从图上看,signature 数量增加到 13 左右时,解释方差已经接近 99%。这通常说明继续增加 signature 数量,收益会开始变小。
signature的绘制
确定一个合适的 signature 数量后,就可以继续做分解和绘图。原文代码里写的是 11,你也可以根据前面 plotNumberSignatures 的结果做自己的取舍。
sigs_nmf = identifySignatures(escc_sca_mm ,
11, nmfDecomposition)
save(escc_sca_mm,sigs_nmf,file = 'escc_denovo_results.Rata')
load(file = 'escc_denovo_results.Rata')
str(sigs_nmf)
library(ggplot2)
pdf("maf.pdf",width=18, height=7)
plotSignatureMap(sigs_nmf) + ggtitle("Somatic Signatures: NMF - Heatmap")
plotSignatures(sigs_nmf, normalize =T) +
ggtitle("Somatic Signatures: NMF - Barchart") +
facet_grid(signature ~ alteration,scales = "free_y")
dev.off()
输出结果示例:
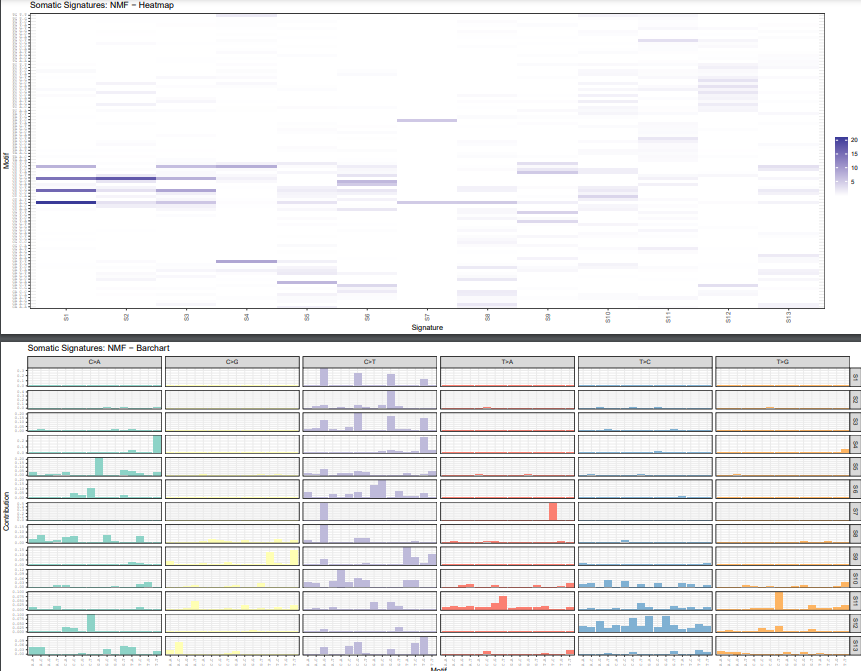
这里通常会得到两类常用图:
plotSignatureMap对应的 heatmap,用来看不同 signature 在样本中的整体分布plotSignatures对应的柱状图,用来看每个 signature 的碱基替换模式
如何快速验证结果是否合理
实际分析里,跑出图并不等于结果就一定可信。至少建议多看下面几项:
- 不同
n_sigs下的解释方差是不是已经趋于平稳 - 各个 signature 是否有明显的生物学模式,而不是纯噪声
- 换一批样本或稍微调整参数后,结果是否还能保持大致稳定
如果你后面还要和 COSMIC 的已知 signature 做比对,也可以把这里的 de novo 结果作为前置探索步骤。
SomaticSignatures 和新一点的工具有什么区别
如果你现在才开始做 mutational signature 分析,通常还会看到另外两个名字:MutationalPatterns 和 SigProfiler。
可以简单这样理解:
SomaticSignatures上手直接,适合快速理解 96-context、NMF 分解和基础可视化流程MutationalPatterns在 Bioconductor 生态里更活跃,扩展分析和与已知 signature 的比较通常更方便SigProfiler在 signature 提取和 COSMIC 体系对接方面更常见,很多新文章也更常引用它
如果你的目标是学习原理、快速做一个可解释的 R 工作流,SomaticSignatures 依然够用;如果你准备做更完整的 signature 拟合、暴露量分析,或者想和更新的公开研究流程保持一致,可以再看 MutationalPatterns 或 SigProfiler。
常见问题
1. mutationContext 报参考基因组相关错误
优先检查染色体命名方式和参考基因组版本是否一致,例如数据是不是 hg38,染色体名是不是 chr1 这种格式。
2. ref 和 alt 一样为什么要提前去掉
因为这类记录本身不构成有效替换,保留下来通常只会在后续构建突变上下文时带来错误或噪声。
3. 为什么选 11 个还是 13 个 signature
这不是固定值,应该结合 plotNumberSignatures 的趋势图来判断。平台期附近通常都值得比较,不必机械照搬一个数字。
总结
本文演示了一条比较实用的流程:先用 Python 清洗 TCGA 的 SNP 数据,再用 SomaticSignatures 构建 96 突变上下文矩阵、评估 signature 数量,并输出 NMF 结果图。
这类方法的价值不在于机械复现某个固定数字,而在于先从自己的样本里做 de novo 探索,再决定是否进一步和 COSMIC 等已知 signature 体系做比较。对于特定肿瘤类型的研究,这通常比直接套用固定模板更有参考意义。
- 原文作者:春江暮客
- 原文链接:https://www.bobobk.com/871.html
- 版权声明:本作品采用 知识共享署名-非商业性使用-禁止演绎 4.0 国际许可协议 进行许可,非商业转载请注明出处(作者,原文链接),商业转载请联系作者获得授权。